Elijah Cole
FMs and Perturbation Prediction: The Role of scRNA-seq FMs
This post discusses results from Foundation Models Improve Perturbation Response Prediction - check out the paper for full details. All opinions are my own.
TLDR: Prior knowledge remains the gold standard, but scRNA-seq FMs prove they can move beyond linear baselines.
Background: Foundation Models for scRNA-seq
Over the last couple decades there has been an explosion of single-cell omics data, especially single-cell RNA sequencing (scRNA-seq). After witnessing the success of foundation models in language and vision, some researchers naturally wondered whether similar foundation models (FMs) could be built for scRNA-seq data. There are many details involved in building these models, but the basic idea is simple enough: Tokenize your scRNA-seq data and pretrain a transformer for next token prediction or masked token prediction or some other self-supervised objective.
What might we want from scRNA-seq FMs? I think the most common answers would be:
- Useful embeddings of genes.
- In particular, contextualized gene embeddings that depend on the cell type or cell state.
- Useful embeddings of cells.
- At a minimum, people like to see clustering according to biological factors (e.g. cell type) and not technical factors (e.g. which lab the data came from).
- Useful counterfactual simulations.
- Example: “If I double the level of gene X, what happens to gene Y?”
scRNA-seq FMs have become a bit controversial. Critics say: “Sure, we can train them - but what exactly are they good for? Are they worth the computational cost?” The paper Deep-learning-based gene perturbation effect prediction does not yet outperform simple linear baselines has been particularly influential in this conversation.
However, our new paper has some nuance to add.
scRNA-seq FMs are not currently the best performers for perturbation prediction
As we highlight in the paper and show in the plots below, the strongest performers are not scRNA-seq FMs. The top 10 methods are all pretrained representations derived from various forms of prior biological knowledge, such as:
- GNNs trained on link prediction using the STRING graph; or
- LLM embeddings of textual descriptions of genes; or
- VAEs trained on gene ontology annotations.
This won’t be surprising to many readers. Curated biological knowledge has been hard-won over decades, and includes lots of useful information about gene function and gene-gene similarity. On the other hand, scRNA-seq FMs are relatively immature as a technology. In particular, most of the work on scRNA-seq FMs could benefit from more rigorous ML methodology, including: stronger baselines, more rigorous ablation studies, and unified benchmark datasets and metrics.
scRNA-seq FMs do perform better than linear baselines for perturbation prediction
Our paper considers the following 7 scRNA-seq FMs:
- AIDO.Cell (3M, 10M, 100M variants)
- Geneformer
- scGPT
- scPRINT
- TranscriptFormer (We also consider many other FMs trained on different data modalities.)
We’ll leave the details to the paper, but the way we use these models is pretty straightforward In brief, we take some control (i.e. non-perturbed) scRNA-seq data, compute gene embeddings for each cell, then average over cells:
gene_embs_per_cell = model(scrna_seq_data) # num_cells x num_genes x emb_dim
gene_embs = gene_embs_per_cell.mean(0) # num_genes x emb_dim
Now, to predict what happens when we knock out a gene we train a simple model (e.g. kNN regression) that takes a gene embedding (corresponding to the gene we’re knocking out) and predicts the post-perturbation expression profile. The goal is to accurately predict the outcome for knockouts that were not observed during training.
So what can we say about the current status of scRNA-seq FMs and their utility for perturbation response prediction? Let’s take a look at how different methods perform on the K562 cell line in the Essential dataset:
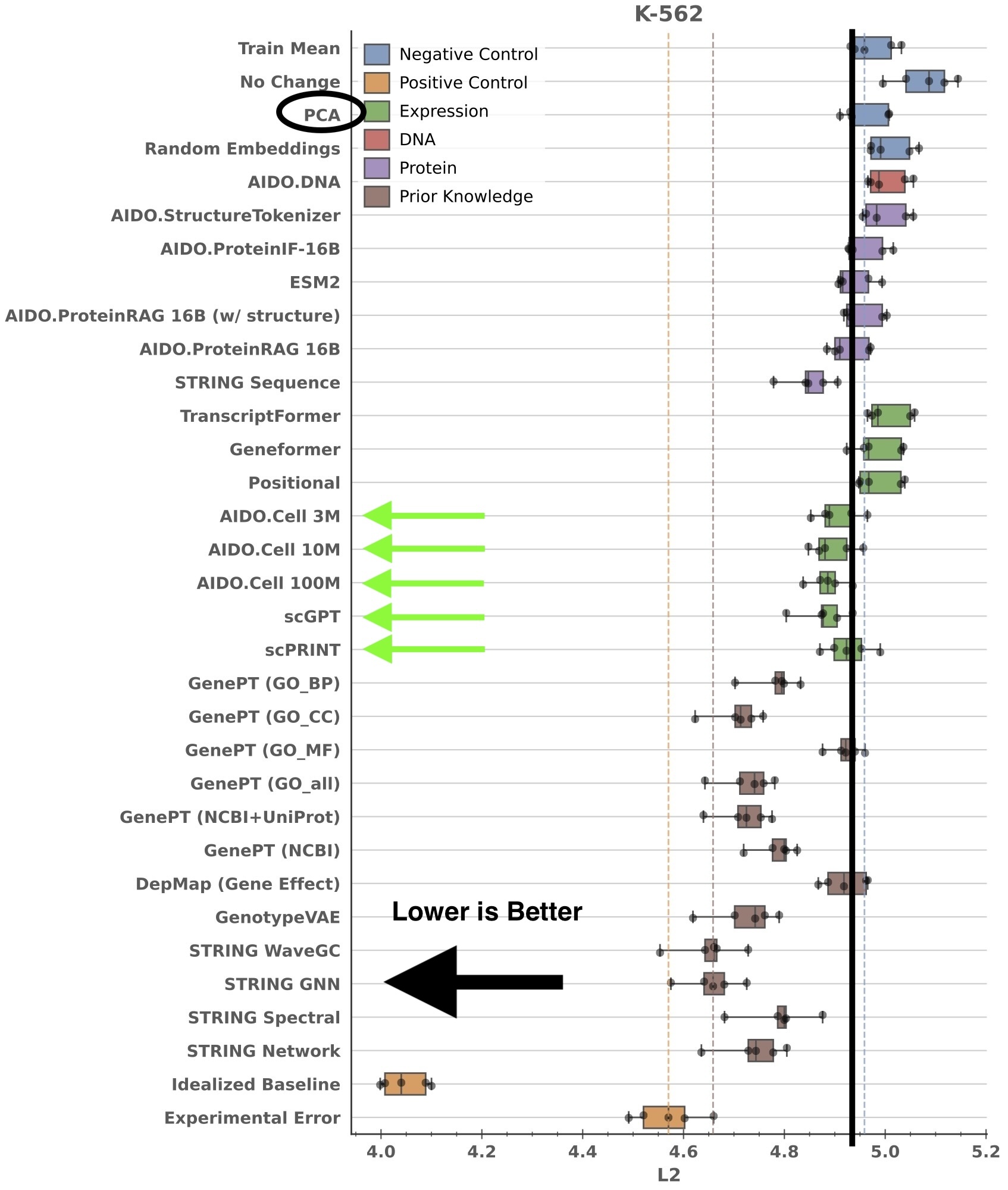
(Note: The error bars in this figure represent the distribution of results of 5 different train/test splits - 5 points per box total.)
We can also look at the distribution of method rankings over all four cell lines in Essential:
![]()
(Note: The error bars in this figure represent the distribution of results for 5 different train/test splits on each of four cell lines - 20 points ber box total.)
It’s easy to see that scPRINT, AIDO.Cell, and scGPT all perform better than a simple baseline (PCA-based embeddings). One can dispute whether the effect size is large enough to justify the cost of training an scRNA-seq FM, but the oft-repeated claim that “scRNA-seq FMs do not beat linear baselines” is not supported by our results.
Conclusion
Will scRNA-seq FMs revolutionize perturbation prediction in the future? I’d say it’s too early to tell. They do perform better than simple linear baselines, but there is plenty of room for improvement. Better architectures, dataset selection, and training procedures could make a big difference. I’m not convinced that we’ve done a good job exploring the design space yet. However, I think improvement will be slow until reviewers and the community at large start to expect more rigorous ML methodology in scRNA-seq FM papers (e.g. ablation studies and strong baselines).